
1Pediatric Resident, Universidad Libre Seccional Cali, Cali, Colombia.
2Pediatrician. M.Sc, Ph.D, Medical Genetics. Post graduate in Clinical Bio informatics and Genomics, Post graduate
Professor at the Health Faculty, Universidad Libre Seccional Cali, Cali, Colombia.
3Pediatric Research Group (GRINPED), Cali, Colombia.
4Neurogenetic and Metabolic Diseases Research Line, Cali, Colombia.
*Corresponding author : Nathalie Yepes Madrid
Pediatric Specialization Resident, Universidad Libre Seccional Cali, Cali, Colombia.
Email: nathalieyepesmadrid@yahoo.com
Received: Sep 10, 2024
Accepted: Oct 18, 2024
Published Online: Oct 25, 2024
Journal: Journal of Artificial Intelligence & Robotics
Copyright: © Yepes Madrid N (2024). This Article is distributed under the terms of Creative Commons Attribution 4.0 International License.
Citation: Yepes Madrid N, Moreno Giraldo LJ. Reverse phenotyping and artificial intelligence in identifying novel and De Novo variants in the RAB39B gene associated with X-linked epilepsy and intellectual disability. J Artif Intell Robot. 2024; 1(2): 1011.
Pediatric epilepsy, which affects 0.5%-1% of children-predominantly those under 5 years of age-is frequently associated with cognitive challenges, drug resistance, and increased mortality. Early-onset cases occur with an incidence of 82.1-118 per 100,000 person-years and are frequently accompanied by developmental delays.
We present a case of a 7-month-old male from a non-consanguineous family with no prior history of epilepsy, who exhibited drug-resistant seizures. Although the electroencephalogram (EEG) findings results were abnormal, magnetic resonance imaging (MRI) findings were unremarkable. Given the suspicion of a genetic etiology, whole-exome sequencing (WES) was performed. Whole-Exome Sequencing (WES) was performed. WES, utilizing DNB-SE for genome-wide coding analysis, focused on exonic regions to detect copy number variants. A novel variant in the RAB39B gene (NM_171998.4), c.434A>G, resulting in p.Tyr145Cys, was identified. This gene is linked to Intellectual Development Disorder type 72 and Waisman syndrome, both of which are X-linked recessive conditions. The variant was absent from major databases, including ClinVar and gnomADv4.1. Initially, classification algorithms like REVEL, BayesDel noAF, and MetaRNN yielded uncertain results. However, subsequent analyses using BayesDel addAF, MVP, and PrimateAI confirmed the variant as pathogenic. Brain-specific expression data from BioGPS further supported the genotype-phenotype correlation.
This variant, located at a splice site, has not been documented in several databases, including NCBI, MedGen, HGMD, OMIM, the 1000 Genomes Project, ExAc, LOVD3, gnomAD, ClinGen, and the Database of Genomic Variants (DGV). High-throughput bioinformatic algorithms (MetaScore, individual predictors) and artificial intelligence tools (GenAI, VarChat, Alphafold, Mastermind, and Alliance of Genome Resources Version 7.1.0) predict a deleterious effect. According to Richards et al. (2015) guidelines from the American College of Medical Genetics and Genomics (ACMG), this variant is classified as probably pathogenic, supported by criteria PVS1, PS3, PM5, PM6, PP3, PM2, and PP2. The early identification of this variant through clinical suspicion and multimodal studies, including omics techniques and AI, facilitates accurate diagnosis and personalized treatment. This approach supports effective genetic counseling, prognosis, and long-term management, use of reverse phenotyping aligning with the principles of 7P Medicine—Precision, Personalized, Predictive, Preventive, Participatory, Proactive, and Population-based care.
Keywords: Gene; RAB39; De novo new; Neuro developmental disorders; Epilepsy; Reverse phenotyping; Multimodal; Artificial intelligence; Precision medicine.
Epilepsy is one of the most common chronic neurological conditions seen in pediatric patients, affecting 0.5%-1% of the population [1]. Its incidence is age-dependent, with the highest rates (>60 per 100,000) found in individuals younger than 5 years old. Several population-based studies have observed a significantly higher incidence of epilepsy in the first year of life compared to older children (82.1-118 vs. 46 per 100,000 person-years) [2,3]. Children with early on-set epilepsy very early in life experience a high burden of cognitive and behavioral comorbidity, and higher rates of drug resistanceand mortality. Up to 50% showing global developmental delay two years after presentation [2,4]. The International League Against Epilepsy (ILAE) Task Force on Nosology and Definitions provides a framework for classifying epilepsy syndromes with onset during the neonatal period and infancy, encompassing infants from birth—whether premature or full-term—up to two years of age. The most recent classification (2022) retains this definition, characterizing epilepsy syndromes as clusters of features defined by typical seizure types, electroencephalogram (EEG) and imaging patterns, age-specific traits, and distinctive comorbidities. These features are essential for determining etiology, prognosis, and treatment strategies [2,4]. Epilepsy is a complex and heterogeneous neurological disorder characterized by recurrent unprovoked seizures. Historically, epilepsy classification has been based primarily on clinical features and EEG findings. However, in recent years, the field of epilepsy research has undergone a profound transformation due to the growing recognition of the significant role genetics plays in the classification of epilepsy subtypes. Currently it’s known that epilepsy has high genetic heterogeneity, in diagnostics, the parallel sequencing of a panel of genes may speed up the determination of the molecular etiology and/or establish a risk of recurrence in family members for the purpose of planning appropriate preventive and/or therapeutic measures. Now many epilepsy syndromes once considered idiopathic or cryptogenic have been linked to specific genetic variant that can be associated with epilepsy subtypes [5,6]. Identifying the etiology of epilepsy is a crucial aspect of epilepsy management, as it aids in prognostic counseling, monitoring comorbidities, and implementing targeted treatment strategies. The advent of advanced diagnostic tools, particularly in genetics, has led to the discovery of many genetically based epilepsies. In contrast to the previous era of single-gene testing, which diagnosed less than 5% of cases, next-generation sequencing provides a genetic diagnosis in 10% or more of patients, with a diagnostic yield approaching 30% in those with underlying epileptic encephalopathy. This increasing identification of genetic epilepsies has shifted treatment practices toward precision-based medicine strategies [1,7]. Reverse phenotyping is particularly advantageous when examining rare phenotypes that fall outside routine assessments, require specialized tests, or involve targeted inquiries into personal and family history. Unlike traditional phenotypic ascertainment research, which begins by grouping individuals based on shared observable characteristics and then seeks genetic explanations, reverse phenotyping starts with the identification of a genetic variant and works backward to identify the associated phenotype. This method allows for more precise examination of conditions that might be missed by conventional approaches due to phenotypic heterogeneity. By focusing on individuals with known genomic variants, reverse phenotyping reduces the risk of cohort selection bias, which can occur when research is confined to specific disease traits or severity levels. Additionally, reverse phenotyping enables the discovery of genotype-phenotype associations that might not fit within the initially defined clinical scope, making it a valuable tool in predictive genomic medicine and genomic screening. This approach has particular relevance for genetic epilepsy research, where uncertain variants like those in the RAB39B gene may lead to novel insights into neurodevelopmental disorders and epilepsy syndromes [8]. Precision medicine is a rapidly growing branch of therapeutics developed on human genetic makeup, lifestyle, gene expression, and the surrounding environment. Researchers can use it to tailor prevention and treatment by identifying the characteristics that expose people to a particular disease and characterizing the primary biological pathways that cause the disorder. Genomic medicine is a relatively new medical specialty that uses genetic information about an individual in treatment for diagnostic or therapeutic purposes and the associated health outcomes and policy implications. Precision and genomic medicine combined with artificial intelligence can potentially improve patient healthcare [9]. Artificial Intelligence (AI) is a powerful approach for solving complex problems in the processing, analysis, and interpretation of omics data, as well as the integration of multi-omics and clinical data. In recent years, AI has enabled remarkable breakthroughs across diverse biomedical fields, such as genomic variant interpretation, protein structure prediction, disease diagnosis, and drug discovery [10].
Genetic alterations, either copy number gain, deletions, or point variants, of the X-linked RAB39B locus, are associated with complex neuropathological clinical features, ranging from X-Linked Intellectual Disability (XLID) in comorbidity with autism spectrum disorders, macrocephaly, and seizures (OMIM: 300774) or with early Parkinson’s disease (OMIM: 311510) [11]. Described RAB39B variants and clinical phenotypes finding that there are seventeen pathogenic variants, confirming that RAB39B downregulation is associated with severe ID/cognitive impairment and significant neurological problems [12].
RAB39B is a neuronal RAB GTPase that controls intracellular vesicular trafficking by switching from an active GTP-bound state to an inactive GDP-bound state, and similar to many other XLID genes, plays a role in a common cellular pathway that controls glutamatergic synapses during development and network establishment [11]. RAB39B switches from an inactive GDP-bound to an active GTP-bound state and drives the intracellular vesicular trafficking of GluA2/GluA3 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) from the Endoplasmic Reticulum (ER) to the Golgi complex, finally orchestrating their postsynaptic surface expression. Either way in literature variants leading to the full absence of RAB39B protein expression or increased dosage levels have been described [12]. The objective is to describe a case involving a patient with a novel and de novo variant in the RAB39B gene, associated with Neurodevelopmental Disorders and Epilepsy syndrome.
A 7-month-old male, born to non-consanguineous parents with no family history of genetic variants or epilepsy syndrome, presented with seizure episodes. His latest episode, occurring as infantile spasms four months after the initial seizure, was characterized by flexion of the lower limbs, deviation of the gaze, and associated crying, despite conventional treatment refractoriness. The physical examination of the patient includes features as macrocephaly, delayed psychomotor. Video electroencephalogram revealed epileptiform activity in the right parietotemporal area, while cerebral Magnetic Resonance Imaging (MRI) with an epilepsy protocol showed normal results. Due to the severity and complexity of the seizure syndrome and the patient’s phenotype, as well as suspicion of a genetic cause, whole-exome sequencing analysis was pursued.
Whole-Exome Sequencing (WES) was performed (covering the entire coding region of the genome) using a state-of-the-art DNB-SE mass sequencer. The process began with obtaining a blood sample and extracting DNA from it. The extracted DNA was then fragmented into smaller pieces. Adaptors, which are short DNA sequences, were added to both ends of each DNA fragment to prepare a library of DNA fragments suitable for sequencing. Biotinylated probes, which are short DNA sequences complementary to the exonic regions of the genome, were used to selectively capture the exons. The probes bound to the exonic regions, and these bound fragments were then separated using streptavidin-coated beads that attached to the biotin on the probes. Sequencing was performed using DNB-SEQ technology, followed by data analysis. From these data, the sequences of candidate genes, based on the patient’s clinical characteristics reported in their medical history, were analyzed. The evaluated genes were analyzed with an average coverage greater than 98% and a minimum depth of 20X. The sequencing results underwent bioinformatic analysis: secondary analysis to assess the quality of the sequencing data, and tertiary analysis to align the sequences, call, and filter variants, meeting specific quality criteria. Variants were analyzed against the hg38 reference genome for annotation and variant calling. The analysis aimed to identify variants in exonic regions or splicing regions (at least 20 bp), small insertions, and deletions. This analysis allows the identification of exonic deletions and duplications (also known as Copy Number Variants, or CNVs) and variants involving large gene regions, which will be reported. Upon identifying a CNV, confirmation is done with a secondary method (MLPA or qPCR), as NGS is an indirect method for its identification. Variants outside the coding region or intron-exon junctions (e.g., variants in promoter regions, enhancers, regulatory regions distant from the exomic region) and special variants including dynamic variants, complex recombination’s, structural gene variants (e.g., inversion rearrangements), low-expression mosaic alterations, and epigenetic effects cannot be identified. Variants with a depth of less than 20x, low allelic ratio, or other characteristics that do not meet the minimum technical thresholds and classified as VUS according to ACMG recommendations were not reported. Pathogenic and likely pathogenic variants under these conditions were confirmed by Sanger sequencing.
This analysis detected, as detailed in Table 1, a variant in the RAB39B gene (NM_171998.3), c.434A>G, resulting in the amino acid substitution p.Tyr145Cys with initial classification of variant with uncertain significance. Pathogenic variant is linked to Intellectual Developmental Disorder type 72 (IDD72), which follows an X-linked recessive mode of inheritance. IDD72 is characterized by intellectual disability, seizures, and macrocephaly. Additionally, this variant has been associated with Waisman syndrome, another X-linked recessive disorder involving intellectual disability and early-onset Parkinsonism. Both conditions are caused by mutations in the RAB39B gene, with overlapping but distinct clinical features.
| Gene | Variants | Zygosity |
|---|---|---|
| RAB39B(NM_171998.3) | c.434A>Gp.Tyr145Cys | Homozygous |
Reanalysis in the geneRAB39B (HGNC: 16499), report two transcripts: NM_171998.4 and ENST00000369454.3 (Exon 2). It is classified as a missense germline variant with Clin Gen Curation ID: CCID: 007740, having sufficient evidence for haploinsufficiency (3) according to PUBMED: 20159109, PUBMED: 21076407, and PUBMED: 23871722. The Triplosensitivity (TS) score is 0. UniProt Q96DA2 · RB39B_HUMAN has an annotation score of 5/5. Genome annotation databases include Ensembl: ENST00000369454.4, ENSP00000358466.3, ENSG00000155961.5; GeneID 116442; KEGG hsa:116442; MANE-Select ENST00000369454.4, ENSP00000358466.3, NM_171998.4, NP_741995.1; UCSC uc004fne.5; Proteomes Identifier UP000005640. Conservation Scores: phyloP100: 5.745. The RAB39B c.434A>G p.Tyr145Cys variant has not been reported in ClinVar or other databases such as gnomAD (v.4.1.0), gnomAD (Aggregated), TOPMed Bravo, 4.7KJPN, GenomeAsia, GME Variome, Iranome, National Center for Biotechnology Information (NCBI) genome, NCBI GenBank, 1000 Genomes Project, Exome Aggregation Consortium (ExAC), Human Gene Variant Database, Leiden Open Variation Database, InterVar-Genetic, nor ANNOVAR browser. Frequencies in exomes: not found (coverage: 29.7), genomes: not found (coverage: 24.1).
The in-silico predictions and meta scores for the identified variant are as follows:
Meta scores with pathogenic significance or VUS
BayesDel addAF: Pathogenic Supporting with a score of 0.1711 (dbNSFP version4.).
BayesDel noAF: Uncertain with a score of 0.007974 (dbNSFP version 4.8).
MetaRNN: Uncertain with a score of 0.5247 (dbNSFP version 4.8).
Individual predictors with pathogenic significance or VUS:
MVP: Pathogenic Moderate with a score of 0.986 (dbNSFP version 4.8)
PrimateAI: Pathogenic Supporting with a score of 0.8676 (dbNSFP version 4.8).
BLOSUM: Uncertain with a score of -6 (BLOSUM100 version).
DANN: Uncertain with a score of 0.9841 (2014 version).
FATHMM: Uncertain with a score of -1.35 (dbNSFP version 4.8).
FATHMM-MKL: Uncertain with a coding score of 0.9679 (dbNSFP version 4.8).
LRT: Uncertain with a score of 0.000011 (dbNSFP version 4.8).
M-CAP: Uncertain with a score of 0.09798 (dbNSFP version 4.8).
MutPred: Uncertain with a score of 0.533 (dbNSFP version 4.8).
In summary, the BayesDel addAF and PrimateAI predictors indicate pathogenic significance, while several other scores suggest uncertain significance, highlighting the complexity of interpreting this variant.
Franklin by Genoox suggests a classification of VUS (Variant of Uncertain Significance).
UCSC Genome Browser on Human Genome Location: chrX: 155258235-155264491 (hg38).
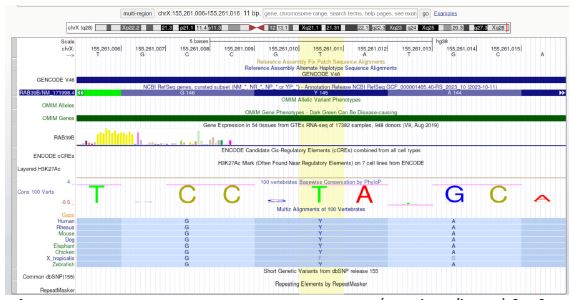
Molecular function: GTPase activity (GO:0003924), protein binding (GO:0005515), GTP binding (GO:0005525), myosin V binding (GO:0031489). Biological process: Autophagy (GO:0006914), regulation of autophagy (GO:0010506), protein transport (GO:0015031), vesicle-mediated transport (GO:0016192), Rab protein signal transduction (GO:0032482), synapse organization (GO:0050808). Cellular component: Golgi apparatus (GO:0005794), plasma membrane (GO:0005886), cytoplasmic vesicle membrane (GO:0030659), vesicle (GO:0031982), neuron projection (GO:0043005). Interpro: Small GTPase (IPR001806), Small GTP-binding protein domain (IPR005225), P-loop containing nucleoside triphosphate hydrolase (IPR027417), Rab39 (IPR041818).
According to artificial intelligence tools, VarChat characterizes the c.434A>G variant in the RAB39B gene as a missense variant that leads to an amino acid substitution from tyrosine (p.Tyr145Cys). This single nucleotide change modifies the codon from TAC to TGC, replacing a polar, aromatic amino acid with a smaller, polar, and sulfhydryl-containing one. This alteration could impact the protein’s structure and function, as cysteine has the potential to form disulfide bonds, potentially affecting the protein’s tertiary or quaternary structure. Mastermind has not yet provided an assessment for this variant, while Genome GPT notes that RAB39B, a member of the RAB family of small GTPases, plays a crucial role in vesicle trafficking regulation. The p.Tyr145Cys substitution occurs at a conserved position, which might significantly affect the protein’s function (NCBI). The structural change from tyrosine to cysteine could potentially alter the protein’s stability and function, leading to clinical manifestations in affected individuals. In-silico analysis provides inconsistent predictions regarding the impact of the p.Tyr145Cys variant. However, the conservation of the tyrosine residue across species and the pathogenic nature of other variants at this position support the variant’s potential clinical significance (NCBI). Population data suggest that this variant is rare and not commonly found in large databases, indicating it is likely a rare variant with potential pathogenic effects (NCBI).
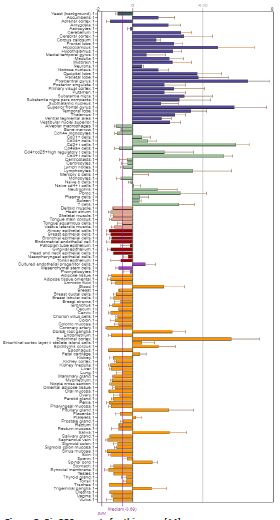
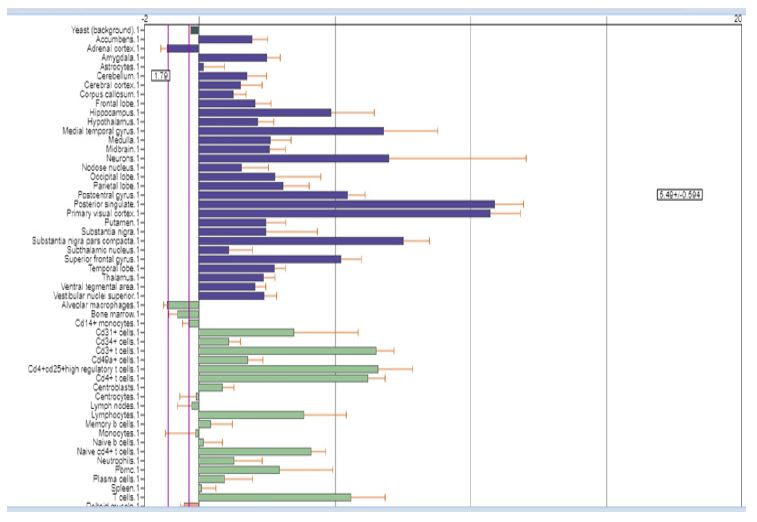
The AlphaFold protein structure database predicts sequences found in UniProt reference proteomes. RAB39B is involved in regulating intracellular membrane trafficking, which is essential for vesicle formation, movement, tethering, and fusion (PubMed: 27103069). It also regulates SNCA/alpha-synuclein homeostasis and, along with PICK1, facilitates the selective exit of GRIA2 from the endoplasmic reticulum to the Golgi, affecting synaptic transmission. According to the Human Phenotype Ontology (HPO) platform, the gene RAB39B NCBIGene: 116442, also known by synonyms as BGMR, MRX72, WSMN, WSN, and XLID72, is located on Xq28 and encodes a member of the Rab family of proteins. Rab proteins are small GTPases involved in vesicular trafficking. Mutations in this gene are associated with X-linked cognitive disability. This gene is linked to several conditions, including early-onset Parkinsonism-Intellectual Disability Syndrome, also known as Waisman Syndrome (OMIM: 311510, ORPHA: 2379), which is characterized by macrocephaly, frontal bossing, delayed psychomotor development, mental retardation, poor speech, and Parkinsonism (L-DOPA responsive). Other symptoms may include tremor, rigidity, shuffling gait, bradykinesia, dyskinesia, dysarthria, dementia (in some patients), seizures (in some patients), loss of dopaminergic neurons in the substantia nigra, and the presence of alpha-synuclein-immunoreactive Lewy bodies and neurites, tau immunoreactivity, and axonal spheroids. And also Intellectual Developmental Disorder, X-linked 72 (OMIM: 300271), characterized by developmental delay, dolichocephaly, macrocephaly, a long face, and mental retardation ranging from mild to severe. Other features may include seizures, limited memory, attention, and language, as well as behavioral and psychiatric manifestations such as stereotypic behavior, hyperactivity, and, rarely, autistic features.Motor skills in affected individuals are generally less impacted compared to cognitive skills.
| Gene | Location | Pathology | Definition | MIMnumber | Inheritance | Synonyms | DiseaseAssociations | |
|---|---|---|---|---|---|---|---|---|
| ORPHA | OMIM | |||||||
| RAB39B | Xq28 | Waisman syndrome withX- linked recessive inheritanceor Early-onset parkinson- ism-intellectual disability syndrome with X-linked recessive inheritance | A rare X-linked syndromic intellectual disability characterized by infantile-onset non-progres- sive intellectual deficit (withpsychomotor de- velopmental delay,cognitive impairment and macrocephaly)and early-onset parkinsonism (before45 years of age), in male patients. | 300774 | X-linked recessive | Laxova-Opitz syndrome | 2379 | 311510 |
| Waisman syndrome | ||||||||
| Mental retardation,X-linked 72 withX-linked recessive inheritance | is a prevalent neurodevelopmental disorder | 300774 | X-linked recessive | MRX72, intellectual developmental disorder, X-linked 72, X-linked recessive, intellectual disability,X-linked 72, intellectual disability,X-linked type 72,mental retardation, X-linked 72, mental retardation,X-linked type 72 | 777 | 300271 | ||
| X-linked intellectual disability(XLID) affects 1 to 3% of thepopulation (Bhasin et al., 2006; Larson et al., 2001;van Bokhoven and Kramer, 2010).Clinically, XLID is characterized by a defi-cit in intellectual function with an intelligence quotient (IQ) of < 70 before the ageof 18 and impairment of adaptivebehaviors leading to deficient communication and social interaction | ||||||||
According to population databases, the c.434A>G variant in the RAB39B gene is classified as pathogenic with a moderate significance. It is noted for its extremely low frequency in gnomAD v4.1 and other previously described databases. The variant has a PVS1 classification for its effect on the protein, indicating a very strong pathogenic significance. This classification is supported by the fact that it occurs in a gene where loss of function is a known disease mechanism. ClinVar reports eight pathogenic null variants for this gene, including several specific changes in the same genomic region chrX:155260886:C>A:HG38, chrX:155260885:TC>T:HG38,chrX:155264166:CACGGTGGGGTCAGAAACCTGGGCAAAGCGACCCTCGGTGAAGCGGCGGATCAGGCAGGACTTGCCCACTGTGGAATCCCCGATGACAATGAGCCGGAACTGGTACAGCCAGATGGCCTCCATGGCCGCGGCTCTGCAGGTCTCCTTGGCCCGGACCGGGGACGGCGGGAGGGGGCGCCCCGCCGACGCCTCAGAGAGCGCGGCTCGGCAGGATCTAGCTCAGCCGCGAGCGCATCGCTGGCCTGGGAGCCCGAAGCTGGGTAGGCCCAGGCGCCGGGAGAGCGGAGGGATGGATGCTGCGCTCGCAAAGGTGATGAAATGGCAGCAGCATCAGCACCACGCACTCTGACTCATCACTGACAACCGGGTTACTGTAATCCCCCGCGTAGACGCGACGCCTGGGCCGCCGCACTATCTGTAAGAGGAGCGCACACAAAATGGCTGCAACATTAGCGCGATCCCTCCCTTTCTAACTACCCCACCCACCCTCTGTTCATTAAAACTGTTCTCTGTCGTTCAAACTGCCAATGTCATACCTTTTCATTGAAACTAGCAAAGCAATACAAAACTGCACAAAAAAGGGAAGGCACATAATTATATATTAGTCTACAAGTGTTTCCATGCTCACATAAACACACAAATATTATATGTGTTTGTATTCCTATATGCATTTATTGCTGTTTGTTTTTACAAAAATTAAACTGGGGCCTTACTTTCAATTAATAATTGCTTTTTGCAATTAATAAAGCAACATAAATATCTTTCCATTTCCACCTAATTTCCCTTTTGTCATAACTTATTTTAAAGTGTAGCCACAAGTATTTTATTTCTGTGAGCTACAGTCTTAAAAAAGGGGCCATGAATTCAAAGAGTATAAGTATTTTAAATTTAACGGACATGGGCATTCTACTTTCCAAAACTATTTAACAGTTTAAATTCTACCCAGCAATGTGTGAAAAGACCATCGCTCTGCATTATTTCAAGCATCATATGTATGTTTTATTCTTTCTTAATTCTTCTCAATAGAATTTAGTTAAATAAAAATATGTCTCATTGCTTTATTTTTACATTTCTCTGACTACTGATGAGGTCAATACCAAGTCTTGTGTATTGACCATTTCATTTCCTCTTCTGGGAATTGTCTCTTCATATCCTTTGTATTTTTTATGTGTAATTTTATTTTATTTTTTTTTTGAGACAGAGTCTCTCTCTGTCGCCCAGGCTGGAGTGCAGTGGCGCGATCTCGGCCCACTGCAACCTCTACCTCCCGGGTTCATGCCATTCTCCTGCCTCAGCCTCCCGAGTAGGTGGGACTACAGGTGCCCGCCACCACGCCCGGCTAATTTTTTGTATTTTTAGTAGAGATAAGGTTACACTGTA>C:HG38, chrX:155264101:C>T:HG38.), which span two different exons and include two variants within this exon. The gnomAD observed/expected score is 0.52. The variant has been classified as PS1 (Pathogenic Strong) because it has not been previously identified or recognized in ClinVar or by other databases, including Genoox and UniProt. It is also classified under PM4 and PM5 (Pathogenic Moderate) due to the protein length changes resulting from a non-repeat region and different amino acid changes that are known to be pathogenic. Furthermore, it has a PM1 classification (Pathogenic Moderate) as it is a non-truncating non-synonymous variant located in a mutational hot spot. In terms of de novo data (PM6), this variant is considered pathogenic with moderate significance as it appears de novo in a patient with a consistent phenotype and no family history, with both maternity and paternity assumed. Overall, the variant is classified with PVS1, PS1, PM4, PM5, PM1, PM6, PP3, and PM2. This classification supports a genotype-phenotype correlation, utilizing ontological platforms such as HPO (Human Phenotype Ontology), OMIM (Online Mendelian Inheritance in Man), Gene Ontology (GO), and ORPHANET. The variant is not reported in ClinVar, by the Genoox community, or in the UniProt database and is noted for its extremely low frequency in population databases, indicating a novel, potentially pathogenic variant.
RAB39B, a member of the RAS oncogene family in Homo sapiens, encodes a protein involved in vesicular trafficking as a small GTPase. Variants in this gene are associated with X-linked cognitive disability. Cheng et al. first cloned RAB39B during large-scale sequencing of a human fetal brain cDNA library. The 213-amino acid protein, with a molecular mass of 24 kD, features 4 GTP/GDP binding domains, 5 RabF domains for regulatory interactions, and a C-terminal prenylation motif (xxCxC). Northern blot analysis showed expression of RAB39B in 16 human tissues, except heart and liver; PCR analysis confirmed its presence in all other tissues [15]. RAB39B is located on the X chromosome (Xq28) and encodes a protein that belongs to the Rab family of small GTPases, which are critical in vesicle trafficking. Rab proteins switch between GTP-bound active and GDP-bound inactive states to regulate intracellular vesicle transport. Recent studies have linked loss-of-function variants in RAB39B to several diseases, including X-Linked Intellectual Disability (XLID), Autism Spectrum Disorder (ASD), and Parkinson’s Disease (PD). Despite this, the exact physiological functions and pathological roles of RAB39B remain unclear [16]. Variants in RAB39B (MIM 300774) at Xq28 cause a syndromic form of X-Linked Intellectual Disability (XLID), with a few affected males documented (MRX72; MIM 300271). Patients exhibit diverse neurological symptoms, including moderate to severe Intellectual Disability (ID), seizures, Autism Spectrum Disorder (ASD), macrocephaly, delayed psychomotor development, and early-onset Parkinson’s disease, akin to Waisman Syndrome (WSMN; OMIM 311510) [17]. Notably, RAB39B has been associated with a range of phenotypic outcomes, reflecting variability in the clinical presentation of XLID, as well as potential links to other conditions such as early-onset Parkinson’s disease. The human RAB39B gene, which maps to the distal Xq28 locus, encodes RAB39B, a member of the RAB GTPases, small monomeric GTPases that belong to the RAS-type GTPase superfamily, which have a key role in the regulation of intracellular vesicular trafficking. Indeed, RAB GTPases are physically associated with specific organelles, through the interaction of specific effector proteins, and act as a network to both spatially and temporally regulate the transport of specific vesicles that switch from the active GTP-bound state to the active state. Inactive attached to GDP. RAB and RAB-associated proteins have been shown to play an important role in a number of rare monogenic and multifactorial diseases characterized by cognitive deficits. In particular, previous work on X-Linked Intellectual Disability (XLID) has identified variants in the GDI1 gene that cause loss of function of the RAB-interacting protein αGDI. RAB GTPases and interacting proteins are abundantly expressed in the central nervous system in all species, and Gdi1 mouse models have been relevant to demonstrate the causal link of loss of αGDI function with deficits in glutamate release and cognitive impairment. One group previously demonstrated that loss-of-function variants in the human RAB39B gene are associated with XLID comorbidity with autism spectrum disorder and seizures and also comorbidity with early-onset Parkinson’s disease. Furthermore, a tandem copy number gain of 0.5 Mb in distal Xq28, including RAB39B, was also reported to be linked to XLID. Therefore, studies define that RAB39B is a specific neuronal protein located in the Golgi compartment, but its functional role is unknown. The demonstrated link to a human genetic disease prompted us to define the functional role of RAB39B in neurons and the potential link to cognitive function [11,18]. RAB39B, together with PICK1, drives trafficking of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) GluA2/GluA3 along the secretory pathway to achieve insertion of the AMPAR subunit into the neuronal glutamatergic postsynaptic system. Terminal. Downregulation of RAB39B in primary hippocampal neurons alters GluA2/GluA3 AMPAR trafficking from the Endoplasmic Reticulum (ER) to the Golgi apparatus, affecting AMPAR subunit composition as a final step and leading to increased AMPAR surface expression. Ca 2+ permeable lacking Glu A2 [11]. The subunit composition of AMPARs is very dynamic and is modified during development, in cases of plasticity and by disease. The increase in surface expression of the Ca 2+ -permeable AMPAR composition lacking GluA2 is mainly observed following plasticity-inducing neuronal activity. Pathological conditions have been associated with immature synapses and cognitive impairment [11]. Mignogna et al. on 2022 reported a nonstop variant in the RAB39B gene that abolishes the canonical stop codon, resulting in an extended reading frame with 21 codons of unknown function. As is usual for XLID, the clinical features associated with RAB39B variants are variable and difficult to predict. After their initial description of two families with RAB39B variants and moderate to severe ID associated with macrocephaly (family X, 6 cases), seizures (family MRX72, 3 cases), and ASD (family MRX72, 1 case), only a few other cases have been reported in the literature [12]. Wilson et al. on 2014 reported three brothers with RAB39B deletion, severe ID, and typical parkinsonism that developed in one of them, adding RAB39B-related disease to the group of synucleopathies. A role of RAB39B in parkinsonism has been subsequently described in several cases, usually, but not exclusively, in patients with RAB39B deletions/loss-of-function variants [19]. An increased RAB39B dosage seems to be the most likely candidate for the neurodevelopmental delay and neurobehavioural disturbances of Xq28 duplication syndrome. Despite this evidence, no clear RAB39B genotype-phenotype correlations can be established (Table 2). This could be due to the limited cases reported in the literature or, more likely, to the influence of other genetic and/or environmental factors. Therefore, in addition to identifying new cases, their complete and accurate clinical characterization is very important, (Table 2) [12]. Also the graphical view of RAB39Bwith its functional domains and published pathogenic variants [17].
| Pathogenic variants | Molecular Consequences | Clinical Phenotypes |
|---|---|---|
| Point variants | ||
| c.21C >A; p.Y7X | Nonsense variant; no protein | ID, ASD, epilepsy, macrocephaly |
| c.215 + 1G >A | 5’splice site variant; no protein | ID, ASD, epilepsy, macrocephaly |
| c.503C >A; p.T168K | Missense variant; down regulation | ID, early-onset PD |
| c.574G >A; p.G192R | Missense variant; altered intracellular localization | PD |
| c.428C >G; p.A143G | Missense variant; ND | PD |
| c.557G >A; p.T186X | Nonsense variant; no protein | ID, early-onset PD |
| c.559G > T; p.E187X | Nonsense variant; no protein | ID, ASD,macrocephaly |
| c.640 T >C; p.*214Gext*21 | Nonstop variant; down regulation | ID, ASD,motor problems |
| Deletion/duplication | ||
| c.624_626delGAG; p.R209del | Deletion; ND | PD |
| c.432delA; p.T145Tfs*3 | Deletion; no protein | ID, early-onset PD |
| c.258_260delTCT; p.L88del | Deletion; ND | ID |
| c.436_447del;p.G146_Y149del | In-frame deletion; ND | ID, ASD, epilepsy, macrocephaly |
| c.536dupA; p.E179fs*48 | Duplication; ND | Juvenile PD |
| c.137dupT; p.S47Lfs*44 | Duplication; no protein | ID, early-onset PD |
| c.371delA; p.K124Sfs*10 | Deletion; no protein | ID, early-onset PD |
| Genomic deletion/duplication | ||
| 0.5 Mb dul Xq28 | Copy-number gain;over expression | ID, behavioural problems, speech impairment |
| 45 kb del | Gene deletion; no protein | ID, early-onset PD |
| ND:RAB39B protein characterization not determined; ID: Intellectual Disability; ASD: Autism Spectrum Disorder; PD: Parkinson’s Disease; §: variantdescribed in the presentpaper. | ||
In mouse brain, RAB39Bis expressed in cortical and hippocampal neurons, as well as in dopaminergic neurons of the substantia nigra, concordant with its association with parkinsonism and cognitive impairment in humans. RAB39B knockout mice showed reduced cortical neurogenesis, macrocephaly, and autistic behaviors, similarly to patients with variants in RAB39B. The Ras-related protein RAB39B is a small neuron-specific GTPase that contributes to synapse formation and maintenance by regulating organization and dynamics of intracellular membranes and vesicular membrane traffic. With its effector, the protein interacting with C-kinase 1, RAB39Bregulates availability of AMPA receptor, important for synaptic plasticity. Furthermore, the complex formed by C9orf72, WDR41, and SMCR8 was found to act as a GDP/GTP exchange factor for Rab-8A and Rab-39B, suggesting that Rab-39B might be involved in autophagy regulation. However, precisely how RAB39B loss-of-function or increased dosage can perturb neuronal development leading to cognitive impairment needs further clarification [17].
The concept of reverse phenotyping has emerged as a tool to understand how specific genetic variants relate to observed phenotypes. In the case of RAB39B, reverse phenotyping might elucidate how certain variants lead to diverse neurological outcomes, from intellectual disability to Parkinson’s disease. The variability in clinical presentations among individuals with RAB39B mutations suggests that reverse phenotyping could provide deeper insights into the relationship between genotype and phenotype, potentially highlighting undiscovered links between the gene and its broader effects on cognitive and neurological functions. This approach might also shed light on cases with atypical presentations or unreported phenotypes, contributing to a more comprehensive understanding of RAB39B-related Diseases [8,12,19]. Artificial Intelligence (AI) utilization in health care has grown over the past few years. It also has demonstrated potential in improving the efficiency of diagnosis and treatment. Some types of AI, such as machine learning, allow for the efficient analysis of vast datasets, identifying patterns, and generating key insights. Predictions can then be made for medical diagnosis and personalized treatment recommendations. The use of AI can bypass some conventional limitations associated with rare diseases. Namely, it can optimize traditional randomized control trials, and may eventually reduce costs for drug research and development. Recent advancements have enabled researchers to train models based on large datasets and then fine‐tune these models on smaller datasets typically associated with rare diseases [20]. Technological advances in omics evaluation, bioinformatics and artificial intelligence have made us rethink ways to improve patient outcomes [21]. Reported and discussed the application of Artificial Intelligence (AI) strategies for metabolomics data analysis. Particularly, they focused on widely used non-linear machine learning classifiers, such as ANN, random forest, and Support Vector Machine (SVM) algorithms. A discussion of recent studies and research focused on disease classification, biomarker identification and early diagnosis were presented. Challenges in the implementation of metabolomics-AI systems, limitations thereof and recent tools were also discussed [22].
A variant in RAB39B was classified as probably pathogenic with moderate significance due to its extremely low frequency in population databases, such as gnomAD v4.1. The variant has been classified with PVS1 due to its impact on protein function, with evidence of pathogenic null variants in ClinVar. The variant affects a gene where loss of function is a known disease mechanism, supported by multiple pathogenic null variants reported in ClinVar. Other classifications include PS1 due to the lack of known pathogenic amino acid changes, PM4 and PM5 due to protein length changes and non-repeat regions, and PM6 indicating de novo occurrence in a patient with a consistent phenotype [23]. This extensive classification underscores the genetic complexity of RAB39B-related disorders. AI is a rapidly evolving and dynamic field with the potential to revolutionize various aspects of human life. AI has become increasingly crucial in drug discovery and development. AI enhances decision-making across different disciplines, such as medicinal chemistry, molecular and cell biology, pharmacology, pathology, and clinical practice. In addition to these, AI contributes to patient population selection and stratification. The need for AI in healthcare is evident as it aids in enhancing data accuracy and ensuring the quality care necessary for effective patient treatment. AI is pivotal in improving success rates in clinical practice. The increasing significance of AI in drug discovery, development, and clinical trials is underscored by many scientific publications. Despite the numerous advantages of AI, such as enhancing and advancing Precision Medicine (PM) and remote patient monitoring, unlocking its full potential in healthcare requires addressing fundamental concerns. These concerns include data quality, the lack of well-annotated large datasets, data privacy and safety issues, biases in AI algorithms, legal and ethical challenges, and obstacles related to cost and implementation. Nevertheless, integrating AI in clinical medicine will improve diagnostic accuracy and treatment outcomes, contribute to more efficient healthcare delivery, reduce costs, and facilitate better patient experiences, making healthcare more sustainable [24].
Considering the crucial role of RAB39B in neuronal function and its involvement in various neurological disorders, including epilepsy, we have classified the associated variant based on an extensive review of bibliographic searches, clinical and biological databases, genomic annotations, and recent proteomics and functional studies. The tools and standards used include Genome GPT, VarChat, Alphafold, Mastermind, and the Alliance of Genome Resources Version: 7.1.0, along with guidelines from Richards et al. (2015) and various interpretation algorithms.
The variant in RAB39B has been classified as probably pathogenic. This classification is supported by several factors: the variant is present at an extremely low frequency in gnomAD and other population databases, which meets the PM2 criterion. In-silico predictions did not fulfill the PP3 and BP4 criteria, while functional data did not meet the PM1 criterion for non-truncating non-synonymous variants in critical functional domains. The variant is also classified under PP2 for its presence as a missense variant in a gene with a low rate of benign missense variants, where such variants are a common disease mechanism. Additionally, de novo data criteria (PM6) were unmet. The classification highlights the correlation between genotype, endotype, and phenotype, with significant phenotypic variability observed across various ontological platforms including HPO (Human Phenotype Ontology), OMIM (Online Mendelian Inheritance in Man), Gene Ontology (GO), and ORPHANET. The integration of multimodal studies that utilize omics techniques and artificial intelligence, following current variant interpretation recommendations, enables precise diagnoses, targeted treatments, genetic counseling, monitoring, and prognosis. This approach aligns with the principles of reverse phenotyping and 7P Medicine (Precision, Personalized, Predictive, Preventive, Participatory, Proactive, and Population-based medicine). The identification of the RAB39B variant’s role in epilepsy underscores the importance of continued research into its functional implications and the potential for developing targeted therapeutic strategies based on detailed genetic and phenotypic information.
Ethics approval and consent to participate: Not applicable.
Consent for publication: Written and oral informed consent was obtained from patient and/or their legally authorized representative (LAR).
Availability of data and materials: All data generated or analysed during this study are included in this published article.
Competing interests: The authors declare that they have no competing interests. This manuscript is not being considered by any other journal.
Funding: No funding was received for the redaction of the case report.
Authors’ contributions: Each author contributed to the redaction, proofreading, and correction of the manuscript. NYM contributed to the research, writing, and proofreading, while LJNM contributed in corrected and adding relevant medical changes to the case. All authors read and approved the final manuscript. All authors participated in the acquisition, analysis, and interpretation of the data. Each author has agreed both to be personally accountable for their contributions and to ensure that questions related to the accuracy or integrity of any part of the work, (even ones in which the author was not personally involved), are appropriately investigated, resolved, and the resolution documented in the literature. All authors read and approved the final manuscript.
Acknowledgments: Not applicable.
Competing interests: The authors declare that they have no competing interests.
